See more posts about EIS and polarization tests
Only 10$ per sample for interpreting of your polarization and EIS results Payment Upon Completion Send your polarization/EIS results...
The corrosion mechanism taking place in an aqueous phase with or without mechanical contact is electrochemical in nature. The electrochemical signal is one of the primary sources of information that relates to behavior in potential, current, and electrical charge of a corroding electrode. It arises from processes that cause corrosion and other electrochemical reactions. In a sliding contact in an ionic electrolyte medium, electrochemistry is more likely to interfere with the tribological behavior of tribocorrosion systems. In recent years, attempts by researchers have been made to control the material loss by electrochemical methods for various engineering systems.
Applied online for monitoring in-situ uniform, localized, galvanic, or more forms of corrosion, such techniques are very convenient means to measure corrosion rate of materials. Such methods can also be used in different ways to evaluate their ability to protect materials (as inhibitors, protective layers, coatings). In this chapter, theoretical and experimental applications, fundamental aspects, limits of the electrochemical techniques for corrosion, and tribocorrosion monitoring are presented. Standards developed, so far, by various standardization organizations are reported. Fundamentals of traditional and advanced corrosion methods are described, focusing on their advantages, i.e. sensitivity to low corrosion rates, short experimental duration, and well-established theoretical understanding.
1. Application of electrochemical techniques for determining corrosion rates
In the section below, practical examples are described of how a number of electrochemical techniques could be used to forecast corrosion or tribocorrosion behavior in practical case studies. The focus is on laboratory tests for rapid corrosion or tribocorrosion tests. The examples do not provide bit-by-bit procedures for screening most or all potentialities. Also, the discussion is not about how to set up and conduct electrochemical corrosion or tribocorrosion experiments. Such information can be readily found in instruction guidelines manual or standard references [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17]. The accent is put on the interest and validity of combination techniques to provide a better understanding of the corrosion process and more reliable predictions.
1.1. Linear polarization resistance (LPR)
The concept of “polarization resistance” has presumably been initiated by Bonhoeffer and Jena in 1951 [18]—a subsequent to Wagner and Traud’s works [19, 20]. In their study of the electrochemical behavior of iron samples of different carbon contents, they found that the slope of the polarization curve, i.e., the rate of potential change E with external current i, at the corrosion potential (or open-circuit potential of a mixed electrode), was low for some iron samples and large for others. Defining this slope as “polarization resistance,” RP, as a result of Lange’s suggestion, it was found that there was an unambiguous correlation between the polarization resistance and the corrosion rate, whereas no correlation was found between the carbon content and the rate of corrosion.
Subsequently, Stern and Geary [21] were the first authors to theoretically establish a linear relationship between the polarization resistance and the corrosion rate based on the kinetics of electrochemical reactions (i.e., corrosion current at open-circuit conditions) and the concept of mixed potential theory, first formulated by Wagner and Traud in 1938 (i.e., parameters of the cathodic and anodic E/i relations) [19]. The advantages and limitations of their method have been discussed in a series of published articles [19, 21, 22], and the linearity of the slope of current-potential plot around the corrosion potential has been verified by experimental evidence, thereby avoiding the problem of large current densities. Their theory has been experimentally supported by other authors [19, 21, 22] for different materials and under a variety of environmental conditions. From the 1960s, plenty of publications [23, 24] reported on the use of the polarization technique, which quickly became one of the main electrochemical techniques routinely adapted to rapid corrosion rate measurements, a condition necessary to its success in industrial monitoring corrosion operations.
For a system in which electrode processes involve a slow reaction step at the electrode surface, the rate of reaction is limited by activation overvoltage; the relationship between the reaction rate, or net current density i, and the driving force for the reaction, or potential E, is given by the Butler-Volmer equation. This equation relates i, for a single electrode process, such as Eq. (1) to E by the formula (2),Fe↔Fe2++2e−Fe↔Fe2++2e− E1i=i0[exp(αnFηRT)−exp((1−α)nFηRT)]=i0[exp(αnF(E−Erev)RT)−exp((1−α)nF(E−Erev)RT)]i=i0expαnFηRT−exp1−αnFηRT=i0expαnFE−ErevRT−exp1−αnFE−ErevRT E2
where η is the overpotential, i0 the exchange current density (rate of either the forward or reverse half-cell reaction) at the equilibrium potential Erev, αthe transfer coefficient (usually close to 0.5, but must be between 0 and 1), and n the number of electrons transferred.
The graphical representation of the Butler-Volmer equation, as shown in Figure 1, is called the polarization curve.

Stern and Geary’s theory [21] is based on a simplified corrosion process assuming that only one anodic reaction and one cathodic reaction are involved during the corrosion process. It is therefore inevitable that erroneous results occur when the corrosion process involves more than one anodic or cathodic reaction. To address this problem, Mansfeld and Oldham [25] presented a modification of the Stern-Geary equation by including more than one oxidation and one reduction reaction in a complicated corrosion process. The current-overpotential relationship at electrodes is set by a number of complex physical and chemical phenomena based on experimental conditions. The reactions occurring at the electrode/electrolyte interface are heterogeneous chemical processes that may involve elementary electron-transfer steps (one or more steps) over the electrochemical double layer, ion-transfer, potential independent or chemical steps, etc.
It is well known that the electrochemistry of corroding metals involves two or more half-cell reactions. Suppose there is a simple corrosion system, such as an iron metal (a corroding working electrode) immersed in a sulfuric acid solution, in addition to Eq. (1), the following half-cell reaction (Eq. (3)) also occurs:H2++2e−↔H2H2++2e−↔H2 E3
The dissolution of Fe takes place in the acid electrolyte. At equilibrium, the total anodic rate is equal to the total cathodic rate. In this case, the net rate of either Fe dissolution or hydrogen evolution can be measured at the electrode potential of the steady-state freely corroding condition. This potential refers to the corrosion potential Ecorr, which lies between the equilibrium potentials of the two individual half-cell reactions. At Ecorr, the net rate corresponds to the uniform corrosion rate, icorr, at free corrosion condition. In such system, the relationship between the overpotential (η, applied potential minus corrosion potential) and the current (flowing between the working electrode and the auxiliary counter electrode) is governed by the fundamental Butler-Volmer equation given as follows:i=i0[exp(αnFηRT)−exp((1−α)nFηRT)]=i0[exp(αnF(E−Ecorr)RT)−exp((1−α)nF(E−Ecorr)RT)]i=i0expαnFηRT−exp1−αnFηRT=i0expαnFE−EcorrRT−exp1−αnFE−EcorrRT E4
In Figure 1, the linear relationship between the polarization resistance and the corrosion rate can be easily illustrated graphically. In the small region near the corrosion potential, Ecorr, only a very small perturbation potential, usually less than ±30 mV (typically ±10 mV), is applied above or below the corrosion potential, yielding a linear relationship between the overpotential (η = E − Ecorr) or the polarization from the corrosion potential and the current. Due to this smooth excitation, the LPR technique is not expected to interfere with corrosion reactions. The slope of that linearized curve (i − E) is defined as the polarization resistance, RP, of a corroding electrode (in ohms cm−2 if the current density is plotted or in ohms if the current is plotted), which is mathematically interpreted asRP=(∂η∂i)|E−Ecorr=0RP=∂η∂iE−Ecorr=0 E5
where i is the current density corresponding to a particular value of E.
The corrosion current, Icorr, can be calculated when the overpotential approaches zero and is related to RP as follows:Icorr=1Rp.babc2.303(ba+bc)Icorr=1Rp.babc2.303ba+bc E6
where ba and bc are the so-called anodic and cathodic Tafel slopes or Tafel parameters, respectively (cf. infra). The corrosion current density, icorr, can thus be calculated from Eq. (6) if RP and Tafel constants (ba and bc) are known.
ASTM G59 describes an experimental procedure required to carry out polarization resistance measurement [10]. In agreement with this standard, the potential should be scanned from −30 mV to +30 mV of the corrosion potential at a rate of 0.167 mV s−1.
Many of the foregoing determined corrosion key parameters are based on empirical observations. As with any empirical method, due to the high number of factors involved in a corrosion or tribocorrosion system (e.g., environmental changes, temperature, pH, reagent as chloride ions, pressure, specimen geometry, test setup configuration, etc.), it is not uncommon to observe that the values of ba, bc, and RP are influenced by these operational parameters and are therefore subject to change. Of significance, the slope generated from the i–E curve around the corrosion potential may not be linear and may or may not be symmetrical in the anodic and cathodic regions. The symmetry of the curve (i–E) at the point of equilibrium or at open-circuit potential is obtained only when ba and bc are equal. These values are required for computing the corrosion current and are usually determined by the Tafel extrapolation method (cf. infra).
It is worthy to note that the measurements of Rp can be derived potentiodynamically or by the method of stepwise potentiostatic polarization or by anodic step pulse method. In the potentiodynamic method, the potential is swept at a constant rate (typically 60 mV/h) from the active (cathodic) direction to the noble (anodic) region passing through the corrosion potential while tracking the current density continuously. More information regarding this method can be found elsewhere [3]. Similarly, in the step pulse method, an applied potential is incremented in steps of ±5 or ± 10 or ± 20 mV, starting from a negative potential moving to a positive potential through the corrosion potential. The value of Rp is determined from the slope of the plot of the potential-current. Prior to the tests, a steady-state corrosion potential is required. The open-circuit potential of the corrosion system is first measured, typically for 1 hour (during which time the corrosion potential of most electrodes is stabilized) or until it reaches a stationary state.
Progress is made through competitive advantages between different measurement techniques, including a rapidity in current measurement (generally rather quickly in a few minutes), where only a lower excitation is required (less than ±30 mV, generally ±10 mV), so that the corrosion rate would not be affected by corrosion reactions, an easy measurement of low corrosion rates (less than 0.1 mil/year (2.5 μm/year), and measurements taken repeatedly, the LPR technique can be considered as a nondestructive technique and used for online corrosion monitoring of uniform corrosion rates useful for the field.
The main drawback of this technique is that the Tafel parameters must be known in advance in order to convert the polarization resistance into the corrosion rate. To tackle this problem, several numerical methods [8, 9, 26, 27, 28] have been proposed to obtain both Tafel parameters and corrosion rate from the same polarization measurement in the vicinity of the corrosion rate. Nevertheless, the success is limited since the Tafel parameters thus determined will not be very accurate, which may compromise the nondestructive nature of the LPR technique. Another disadvantage of the LPR method lays in the fact that it will not work properly in low conductive media. Basically, the LPR technique can only be used to determine uniform corrosion rates; it can hardly provide information about localized corrosion.
1.1.1. Illustrative examples of the application of LPR in corrosion and tribocorrosion systems
A modified electrochemical noise technique, namely, electrochemical emission spectroscopy (EES) [29], offers one of the most convincing examples of the application of the LPR technique in tribocorrosion [30]. Indeed, the analysis of noise data in a potential-current plane shows the transposition of the statistical resistance due to electrochemical noise to the resistance due to linear polarization. Noise resistance is often considered equivalent to the polarization resistance, RP [31, 32, 33]. The noise resistance, RN, calculated using a method proposed by Eden et al. [33], for mild steel passive alloy in 0.05 M H2SO4 (corrosion under activation control), is of the order of 48 Ω without any sliding contact. The LPR measured on this mild steel after EES monitoring is shown in Figure 2a. The comparative value of RP obtained by the LPR technique is 50 Ω. The RNvalue obtained using the EES technique is therefore very close to the RPobtained by the LPR technique. Under tribocorrosion conditions (5 N normal force, 10 Hz sliding frequency, 200 μm peak-to-peak displacement amplitude), the plane plot of the potential-current data under steady-state wear-corrosion regime shows a best-fit line through the data points with a positive slope of 54 Ω (see Figure 2b), which roughly corresponds to RN (48 Ω) or RP (50 Ω in Figure 2a). Notwithstanding, no attempt has been made to relate these resistance measurements with the breakdown (i.e., depassivation) or the buildup of any kind of passive film (i.e., repassivation) on the mild steel surface subjected to either a mechanical stimuli (e.g., active-passive wear track zone area or metastable pit area) or in the absence of wear (free corrosion), characteristic phenomena of localized corrosion.

1.2. Tafel extrapolation method
In 1905, Julius Tafel [34] presented the experimental relationship between the current, I, and the overpotential, η, during an electrocatalytic test of the reduction reaction of hydrogen (i.e., protons to form molecular hydrogen) on a number of electrode metals such as Hg, Sn, Bi, Au, Cu, Ni, and so on:η=a+blogIη=a+blogI E7
where the overpotential η is defined as the difference between the potential of the working electrode, E, and the equilibrium potential.
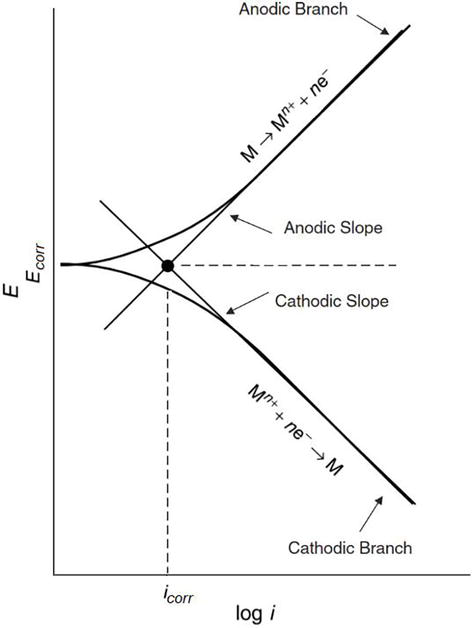
The existence of a linear relationship between E and log I has been demonstrated when the electrode is polarized at sufficiently large potentials, and far away from the corrosion potential both in anodic and cathodic directions [34], as can be seen in the polarization curve depicted in Figure 3. The portions in which such relationships prevail are called Tafel portions or Tafel regions.
This can be mathematically expressed asI=Icorr[exp(2.303ηba)−exp(−2.303ηbc)]=Icorr[exp(2.303(E−Ecorr)ba)−exp(−2.303(E−Ecorr)bc)]I=Icorrexp2.303ηba−exp−2.303ηbc=Icorrexp2.303E−Ecorrba−exp−2.303E−Ecorrbc E8
where Ecorr is the corrosion potential, E the applied potential, η the overpotential (difference between E and Ecorr), I the current, Icorr the corrosion current, and ba and bc are the Tafel constants or Tafel parameters derived from E − log I plots as the anodic and cathodic slopes in the Tafel regions, respectively.
Extrapolating from the Tafel portions of either anodic or cathodic or both, an intersection point is obtained at Ecorr, from which Icorr is readily available from the log I axis. Therefore, it is possible to obtain simultaneously the corrosion current, Icorr, and the Tafel parameters (i.e., ba and bc) from this method.
In order to obtain the Tafel portions in the anodic and cathodic regions, the electrode has to be polarized far away from its corrosion potential, e.g., ±250 mV away from Ecorr. Eq. (8) can be rearranged, as appropriate, to choose one single polarization direction, either anodic or cathodic way.
At sufficiently larger values of η (100 mV ≤ η ≤ 500 mV), in the anodic direction (i.e., η = ηa), Eq. (8) can be rearranged as,ηa=balogIIcorrηa=balogIIcorr E9
Likewise, at sufficiently larger values of η, in the cathodic direction (i.e., η = ηc), Eq. (8) can be modified as,ηc=−bclogIIcorrηc=−bclogIIcorr E10
The polarization curve can be measured either dynamically or statically (either in the potential-controlled mode or in the current-controlled mode). The dynamic polarization techniques can be carried out relatively fast, but the drawback is that the Tafel parameters are scanning rate dependent. The static polarization techniques may produce better Tafel parameters, but they are very time-consuming.
Tafel extrapolation measurements can be performed either by the potentiodynamic method or by the stepwise potentiostatic polarization method [35]. As in RP measurements, in both methods, corrosion potential is first measured, typically for 1 h (during which time corrosion potentials of most electrodes are stabilized) or until it stabilizes. After that, the potential step—at increments of ±25 or ± 50 or ± 100 mV, every 5 min, recording the current at the end of each 5-min period—is applied (potential-step method), or the potential is scanned at a constant rate (typically 0.6 V/h) (potentiodynamic method). In both methods, the experiment is started at the corrosion potential, and the cathodic polarization is first conducted by applying an overpotential of approximately 500 mV or until gas evolution (e.g., hydrogen) occurs at the electrode, at a constant rate of 0.6 V/h. Following, the corrosion potential is measured again (typically for 1 h), and then anodic polarization is conducted by applying an overpotential so that the potential at the end of the anodic polarization reaches +1.6 V versus SCE. Tafel plots are generated by plotting both anodic and cathodic data in a semilog paper as E-log I. From the plot, three values are determined: the anodic Tafel slope, the cathodic Tafel slope, and Icorr (from back-extrapolation of both anodic and cathode curves to Ecorr). The main advantage of this method is that it provides a simple, straightforward method to determine Tafel parameters, namely, ba and bc.
The disadvantage of the Tafel technique is that large current densities are often required to generate the complete Tafel plots. The use of large current densities can alter the surface conditions of the specimen (e.g., permanent change or surface damage), thereby distorting the results and increasing complications due to mass transport and uncompensated electrolyte resistance. The measurement of current density over a wide potential range may also distort the results if the adsorption of some species is potential dependent. Since this method applies a large overpotential to the metal surface (e.g., anodic polarization), therefore, the technique is rather destructive and can hardly be used for online corrosion monitoring purposes and in particular in the field. An ASTM G5 standard provides a procedure for constructing an anodic polarization plot [36]. However, it does not supply a method to construct a cathodic polarization plot nor a procedure to determine the corrosion current by the Tafel extrapolation method.
1.3. Corrosion rate determination by electrochemical noise analysis (ENA)
Many of the electrochemical techniques, among those described earlier, measure the electrochemical response of the corrosion system following the application of an external disturbance. In the last 50 decades, an original concept has emerged where it was possible to use the inherent noise of the electrochemical system as a stimulus to measure both potential and current changes [31, 32, 37, 38, 39, 40, 41, 42, 43]. Broadly, measured inconsistently in corrosion experiments, the electrochemical noise was first considered an unwanted or undesirable artifact that comes from measuring instruments or pickups from the environment. This is why this misleading name was cast. This sort of noise can be easily observed during corrosion potential measurements because the measured corrosion potential always fluctuates slightly, usually randomly. Random fluctuations result from stochastic processes [44], and, considering each chemical process is stochastic in nature, it generates noise.
Since the pioneer work of Iverson [45], who has reported a relation between the frequency or amplitude of the electrochemical noise and the inhibiting power of the environment for a number of metals and alloys (e.g., aluminum alloys, magnesium, mild steel, etc.), there has been a growing interest toward the measurement of electrochemical noise and its peculiar relationship with localized corrosion [11, 12, 31, 32, 41, 46, 47, 48, 49, 50, 51]. In this respect, electrochemical noise measurements obtained from the analysis of corrosion potential or current fluctuations provide a new approach to the study of corrosion processes in reactive environments such as aqueous media or hot aggressive gases or even under the effect of mechanical stimuli, e.g., tribocorrosion. Indeed, mechanical friction of solids in contact with a corrosive environment is likely to generate (i) noise due to stochastic contact between randomly distributed surface asperities and (ii) noise due to the synergy of wear-corrosion processes resulting from the activity of the surfaces and controlled by the response of potential-current transients and the configuration of the wear track area, coordinated by the coupling effects of wear and corrosion in the tribo-electrochemical cell. Among the possibilities offered by the measurement of electrochemical noise sources during an electrochemical or a tribo-electrochemical system, the following can be retained: adsorption–desorption processes, e.g., formation and detachment of gas bubbles; fluctuations in the mass transport rate and in temperature; interfacial nucleation and growth processes; degradation processes due dielectric film disruption; kinetics of atom exchange at the surface sites, e.g., Johnson’s noise in the interfacial impedance; and so on.
While multiple case studies on electrochemical noise have been regularly reported in recent years, even greater progress is possible, with the scope for increased breakthrough in science and technology (e.g., novel materials, precision tools on macro-to-nanoscopic scales, availability and intelligent use of these materials and tools, and so on). In particular, the main focus of these investigations is to promptly obtain in situ mechanistic information on the repassivation and breakdown of passive films and to monitor any process associated with confined corrosion and/or tribo- or bio-tribocorrosion [46, 47, 51]. It has been, indeed, suggested that the noise is caused by film breakdown and repassivation processes, and given the dynamic competition between these two processes, pitting will initiate. However, the foundation for using electrochemical noise analysis for determining the corrosion rate of an electrode is still a subject of debate within the scientific community. Indeed, the fundamental approach is not as robust as that of other techniques. On the other hand, the advantage of the noise analysis is that it is not necessary to apply any external polarization and the system is in natural corrosion conditions. This renders the technique as nondestructive and nonintrusive, capable of monitoring basic changes in an electrochemically active system. This makes it particularly suitable for online corrosion monitoring in the laboratory, especially for localized corrosion monitoring, detection of general corrosion, crevice investigation, stress corrosion cracking [12, 52, 53], fretting corrosion, or be used in the assessment of anti-corrosive organic coatings, and other surface inhomogeneity case studies [43, 46, 47]. Several approaches extend the use of electrochemical noise measurements in both pilot plant and field facilities, its use is not merely limited to the foregoing phenomena, but its development is justified especially when measurements are performed in systems with very low conductivity, where, for e.g., the impedance technique fails because of the loss of signal in the high resistance of the solution (cf. infra).
1.3.1. Instrumentation for electrochemical noise measurements in corrosion and tribocorrosion systems
Electrochemical noise is a generic term used to describe the naturally occurring fluctuations in potential and current, which is due to spontaneous changes in electrode kinetics and mechanisms [33]. When applied to corrosion studies, electrochemical noise may be redefined as the spontaneous fluctuations observed in potential and current at the free corrosion potential. The electrochemical noise can thus be classified into potential noise and current noise. There are three major possible modes for measuring potential and current noise in a corrosion system, but the most common mode uses two nominally identical working electrodes, WE1 and WE2 (WE1 as the corroding metal and WE2 as a counter electrode), and a noise-free reference noble electrode, RE [33] (see Figure 4a). The current flowing between the two working electrodes is measured by a zero-resistance ammeter (ZRA), and their potential is monitored versus the reference electrode through a voltmeter (V) under free corrosion conditions. The two other leftover modes are two identical working electrodes WE1 and WE2 with a bias potential [54] (not shown here) and one WE coupled to a micro-counter electrode (MC, e.g., Pt wire tip) [29, 46, 47, 55] (see Figure 4b). This last mode of electrochemical noise analysis seems to be a promising way to obtain unambiguous estimates of the rate of chemical wear in a tribocorrosion experiment as evidenced by some recent investigations [46, 47, 51] but also to predict the corrosion rate of localized corrosion in a free corroding system [29, 31, 32, 37, 38, 39, 40, 41, 42, 43, 44, 45, 47, 48, 49, 50, 56].

1.3.2. Electrochemical noise data management process
The overall approach to analyzing noise data is the assessment of mechanistic information from either time-domain analysis, frequency-domain analysis, or both, using statistical methods [44, 57, 58]. If the information in the time-domain records is evident, time-domain analysis is sufficient to distinguish different processes (e.g., different forms of corrosion).
In what follows, one assumes that all various types of noise are excluded from this description, with the exception of the thermal noise. Except for the last noise, all other noise sources can be minimized or eliminated using careful strategy within reasonable limits of materiality. Effective and convenient ways include the removal of unwanted environmental and instrumental noise from the electrochemical noise, e.g., by shielding electrical connections/wires for coupling the electrodes to the experimental apparatus, by using a Faraday cage to exclude electrostatic/electromagnetic influences, even by implementing analogue/digital filters to eliminate systematic noise at frequencies different than the frequency of interest, and so on. Guidelines for the calibration of noise measuring device can be found elsewhere [13, 59].
1.3.3. Noise resistance
The basic quantitative approach is the time-domain analysis of the noise signal. The noise resistance, RN, is defined as the ratio of the standard deviations of potential/current noise signal time dependent, σ(t):RN=σE(t)σI(t)RN=σEtσIt E11
Eq. (11) implies that in the case where a low-driving force noise produces a high current density noise between the two electrodes (WE1 and WE2), the yielding noise resistance will be low. Noise resistance, RN, has been shown to correlate well with the polarization resistance, RP, as determined by EIS for certain corrosion systems. This latter being directly related to the corrosion current [14, 29, 60] is using the Stern-Geary equation and Tafel slopes. Notwithstanding, much work has been devoted trying to best match RN or the normalized RN (per unit of exposed surface area) to the corrosion resistance or the corrosion rate [5, 14, 31, 32, 37, 38, 39, 40, 41, 42, 43, 44, 45, 48, 49, 50, 60]. Although signal analysis in the time domain is well established, an approach based on spectral analysis is gaining more and more importance in research laboratories. It consists of transforming the potential and current noise fluctuations recorded in the frequency domain using the Fast Fourier Transform (FFT) method [61].
The frequency range for which the FFT is commonly performed extends from 1 mHz up to 1 Hz. The spectral noise plots are similar to those of impedance plots. The spectral noise resistance, RSN, is given by the ratio of the potential and current FFTs at each frequency, and the limiting value, RSN0, can be used as a measure of the corrosion resistance:RSN(f)=(EFFT(f)IFFT(f))RSNf=EFFTfIFFTf E12
The log–log plot of RSN versus f is similar to the impedance plot, and the spectral noise resistance limit RSN0 is given byR0SN=limf→0RSN(f)RSN0=limf→0RSNf E13
Another approach would be to examine the spectral noise response in terms of power spectral densities (PSD). These latter are calculated from the FFT or using the maximum entropy method (MEM) [62]. RSN is determined from the PSDs by the relation (14):RSN(f)=(EPSD(f)IPSD(f))1/2RSNf=EPSDfIPSDf1/2 E14
It has been shown that the use of a single data set of potential and current noise [32] would yield identical values of RSN as calculated by either Eqs. (13) or (14). In some cases, RSN0 is bound to RN or RP as,RN=RSN(0)=RPRN=RSN0=RP E15
effective if the impedance of the two electrodes is identical and much higher than the resistance of the test solution between them [5, 32, 63]. Experiments have validated this relationship for several systems [5, 31, 63]. Nonetheless, there is no agreement on the fundamental basis for the relationship between noise resistance and corrosion rate.
1.3.4. Illustrative examples of the application of electrochemical noise in tribocorrosion systems
Investigations into electrochemical kinetics make common point research between tribocorrosion and corrosion. The study of localized phenomena of depassivation and repassivation is essential to understand the mechanisms of corrosion-wear as well as to reduce the material loss. The possibility of using the electrochemical noise detection technique as a promising tool to study the electrochemical properties of well-controlled damaged surfaces has been widely considered due to its nondestructive nature and its potential in online corrosion monitoring applications. Time-spatially resolved measurements should provide more reliable data on the electrochemical part of tribocorrosion. The noise analysis in relation to depassivation-repassivation events randomly distributed in time and space can be traced back to Oltra et al. [64]. The power spectral density (PSD) of the noise under the impact of the jet particles was related to the Fourier transform of individual repassivation transients obeying a Poisson distribution. Later, the application of electrochemical noise analysis to tribocorrosion was reviewed. Investigations involving PSD noise analyses on various tribo-electrochemical cells for passivating materials were conducted by Ponthiaux et al. [65], by Déforge et al. [51], and in more details by Berradja et al. [46, 66]. In this latter work, the noise spectra were measured on AISI 304 L stainless steel versus corundum in a Ringer’s solution in a pin-on-disk tribometer under stationary sliding-corrosion regime conditions, either at open-circuit potential or at a controlled potential. The PSD of the current noise has been interpreted as resulting from the overlap of the large number of discrete repassivation transients at the contact junctions, including the double-layer charge and the strong dependence of depassivation and repassivation kinetic rates of the oxide surface film on the sliding frequency. This was consistent with the shift in the PSD plots of the current noise fluctuations by about a decade when the sliding frequency was varied from 0.1 to 1 Hz (see Figure 5). Similar findings were obtained via Déforge et al. [51] by dividing open-circuit potential fluctuations to the impedance of the electrode. A 1/f low-frequency noise is explained by a long-term drift of the surface conditions. Only a minor influence of the applied normal load was observed on the PSD plots, recommending reaching the limit rate of the depassivation of the oxide surface film.

Application of the noise analysis to tribocorrosion offers the feasibility to record in parallel the PSD of normal and tangential force fluctuations and their tie-in with the current noise data (see Figure 6). Force fluctuations show an almost flat spectrum (white noise) as expected following short random mechanical interactions between colliding asperities, whereas the current noise is consistent with finite time-constant transient responses to the depassivation events.

1.4. Corrosion forecast by electrochemical impedance spectroscopy (EIS)
The EIS has matured greatly over the past 25 years as a tool in corrosion protection research and has proven to be one of the most useful electrochemical characterization techniques presently available. In practice, EIS has become a standardized research tool for corrosion prediction [15] and found wide applications in both fundamental and applied laboratory researches [67]. Recent applications in tribocorrosion reflect the steady progress of the EIS method in terms of research and development [62]. Compared with the LPR technique, the EIS technique is considered more advanced, since it has the ability to study high-impedance systems, in which the conventional LPR technique has failed, such as coatings and linings [16, 68], high pure water, and organic coating/metal systems [69] or corrosion in a low conductive solution [70]. This technique is especially useful for evaluating corrosion inhibitors [24, 71], analyzing the corrosion mechanisms [72, 73], and so on.
A significant number of tutorials have been addressed on the EIS experimental setup, the measurement methodology, and data analysis methods [27, 74, 75, 76, 77, 78]. The technique has been of a great deal of concern to the extent that an ASTM standard, i.e., ASTM G106-89, has been produced to provide the practitioner with a test method to verify that the electronic equipment, the electrochemical cell, and the spectrum generation algorithm impedance work properly [15].
1.4.1. Principle of the EIS technique
The EIS technique normally uses a typical three-electrode cell system controlled by a potentiostat, similar to that used in the LPR technique. Unlike the previous time-resolved techniques, where the current system response is either the consequence of a large voltage perturbation from the steady-state condition (case of Tafel extrapolation) or from a smaller perturbation (case of LPR method), in the EIS approach, however, by applying a small varying perturbation over a range of frequency, it is possible to probe the full response of the electrochemical system, and not just the resistive components. In that respect, a small AC signal, i.e., alternating potential or voltage V(ω) typically a sine wave of amplitude ±10 mV of the corrosion potential, is applied over a wide range of frequency (typically from 105 down to 10−2 or 10−3 Hz) at a number of discrete frequencies (typically 5–10 frequencies per decade), and the alternating current response, i(ω), is measured at each frequency, ω (i.e., the ac polarization or angular frequency, ω = 2πf). For a linear system, the current response signal will be a sine wave of the same frequency as the excitation signal (voltage) but shifted in phase. This is transmitted to a frequency response analyzer or a lock-in amplifier to calculate the impedance and phase shift. Full frequency sweeps provide phase-shift information that can be used in combination with equivalent circuit models to gain valuable information from the complex interface of the corrosion system. The frequency-dependent impedance is determined by the relation: Z(ω)=V(ω)/i(ω)Zω=Vω/iω .
1.4.2. Electrode/electrolyte electrochemical interface circuit
Basically, the electrode/electrolyte interface is characterized by a separation of charges resulting in the creation of parallel planes of electrical charges whose behavior is similar to a circuit consisting of a capacitor and a resistor in parallel and certainly not to a perfect capacitor. Indeed, the current flowing in a perfect capacitor would cease when the latter would be fully charged, hence the need to add a resistor in parallel to let a weak current flow. An electrochemical interface can be viewed as an electrical circuit, or called the equivalent circuit, composed of a number of elements such as resistances (R), capacitances (C), and inductances (L) [26]. Explanations of the EIS results are usually based on the equivalent circuit used. Many software programs and packages are now available for fitting the impedance spectra to analogous circuits [15], a strategy often used to analyze data. Further information on the EIS measurements and instrumentation can be found elsewhere [15, 17, 79, 80].
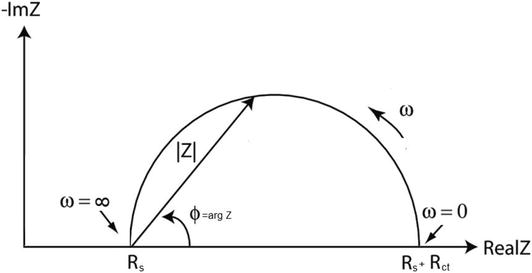
Not all the available proposed equivalent circuits to model electrochemical interfaces can actually satisfy what is applied to a freely corroding system. In most cases, the impedance corresponding to a simple corrosion process, under activation control, can be represented by the well-known Randles’ [81] equivalent circuit (RC circuit) which allows to describe the behavior of many electrochemical electrode/electrolyte interfaces. A typical example is shown in Figure 7, where RS, RCT, and CDL represent labels for the solution resistor, the Faradaic charge transfer resistor, and the double-layer capacitance, respectively. The capacity is associated with the separation of charges at the electrode/electrolyte interface as in the case of a working electrode having a surface film (e.g., AISI 304 stainless steel immersed in a 0.5 M H2SO4 electrolyte), in which case the capacity of the equivalent circuit can be associated with the capacity of the passive oxide surface film and the resistor in parallel with the capacitor is considered as the charge transfer resistance, RCT (or the polarization resistance, RP, under EIS-free corrosion conditions), while the ohmic resistance in solution, RS, between the working electrode and the reference electrode is in series with the parallel resistor and the capacitor. If the amplitude of the perturbation signal is small enough (e.g., a voltage less than 20 mV), RCT can be regarded as equivalent to the linear polarization resistance (RP).

The behavior of such an electrochemical interface can be described by Eq. (16):Z(ω)=Rs+Rp1+(jωRPCDL)βZω=Rs+Rp1+jωRPCDLβ E16
RCT or RP can be determined in several ways. A convenient way is to use the Nyquist diagram. For the simple Randles-type equivalent circuit as shown in Figure 7, the corresponding Nyquist diagram is displayed in Figure 8, in which a perfect semicircle is observed. The high-frequency response is used to determine the component of RS involved in the measurement. RS can be read directly from the abscissa when the angular frequency ω (ω = 2πf) tends to be infinite (fmax or f → ∞). The total resistance (RP + RS) can also be read from the abscissa when ω approaches zero (fmin or f → 0). So, RP can be determined by subtracting the RS value from the low-frequency measurement. The conversion of the polarization resistance into a corrosion rate requires an independent empirical measurement of the Tafel slopes using a potentiodynamic polarization method and/or harmonic distortion analysis or otherwise taken from the literature. The double-layer capacitance, CDL, can also be determined for a system exhibiting a behavior similar to that of a perfect RC circuit from the values of RP and the maximum frequency, fmax, that corresponds to the frequency of the point at which the imaginary component has a maximum value, viz.:

CDL=12πfmaxRpCDL=12πfmaxRp E17
It is worth of note that in practice, f cannot really go as high as infinite; it is inevitable that some extrapolation has to be made. Extrapolation at the high-frequency limit usually presents few issues because the impedance becomes nonreactive at frequencies as low as 10 kHz in most cases [82]. On the other hand, reactance is still commonly observed at frequencies as low as 10−3 Hz [82]. Therefore, special precautions must be taken to obtain reliable data and to avoid possible artifacts [17, 83]. Furthermore, the measurement cycle time depends on the frequency range used, in particular the low frequencies. For instance, a single-frequency cycle at 10−3 Hz needs about 15 min of testing time. A high-to-low-frequency analysis moving down to 10−3 Hz frequency likely requires more than 2 hours of scan time. In order to perform a normal standard corrosion monitoring with the EIS technique, assistance is needed to optimize the use of the high-frequency data and reduce measurement time. There is a constant need to improve data processing and analysis in order to minimize uncertainties and to allow the EIS technique becoming user-friendly for corrosion monitoring in both laboratory and field facilities, though it must be emphasized that the need for an easy-to-deploy field instrument has always been an obstacle to online corrosion monitoring with the EIS technique.
An alternative to the impedance model in the Nyquist diagram involves the conversion of the impedance into a complex number. The impedance can thus be designated by an amplitude, |Z|, and a phase shift, ϕ, or by the sum of the real (Z′) and imaginary (Z″) components, such that,Z(ω)=Z'(ω)+jZ”(ω)Zω=Z′ω+jZ”ω E18
Both the log|Z| data and the phase angle ϕ are plotted against the angular frequency, log ω, of the excitation signal, a format which substitutes for the Nyquist diagram, i.e., the so-called Bode diagram. Figure 9 shows how the same data (Nyquist plot) appears in a Bode plot format with respect to the equivalent circuit of Figure 7.

Highest (ωH) and lowest frequencies (ωL) can be readily determined. As shown in Figure 9, Z is independent of the frequency at ωH and ωL, limit values represented by horizontal lines. From these lines, values of RS and (RS + RCT) can be measured. This analysis forms the basis of the corrosion monitoring as proposed by Tsuru et al. [74] to allow the determination of |Z| at each frequency in the horizontal portions of the Bode diagram.
Sometimes, it is not convenient to perform impedance measurements at very low frequencies (as in DC techniques such as linear polarization). However, it is still possible to extrapolate the polarization resistance, RP, from the Bode diagram. In Figure 9, the low- and high-frequency breakpoints (i.e., ωL and ωH, respectively) can be determined from the 45° phase angle Bode diagram (see the pseudo-Gaussian curve). The intersection point A can be determined from the log ωH and RS. By extrapolating from A toward the central linear portion of the |Z| curve, a linear line can be determined. On this line, point B is obtained at log ωL.With the projection of point B to the log|Z| axis, the total resistance (RS + RCT) can be measured. In this way, RP can be determined. At intermediate frequencies, the capacitor affects the response of the overall RC circuit.
The situation struggles when diffusion processes govern the corrosion behavior. A convenient way to deal with this complication is to add a Warburg impedance. The latter describes the impedance of the concentration and diffusion processes in the equivalent circuit as shown in Figure 10.

The Warburg impedance, ZW, is given by the equationZW=σwω√−jσwω√ZW=σwω−jσwω E19
where σw is the Warburg coefficient.
Eq. (19) implies that, whatever the frequency, the real and imaginary parts of the Warburg impedance are equal and inversely proportional to σw½. In the Nyquist plot, this impedance will result in a straight line at a constant phase angle at 45°, as shown in Figure 10. However, the effect of the Warburg impedance can complicate the correct estimate of the RP value in certain cases. Therefore, the impedance data must be numerically adjusted to fit with the correct model to facilitate the extraction of the total resistance (RS + RP) from the abscissa or by using an appropriate modeling software. However, the situation can readily become more complicated if other effects, such as time-constant dispersion, adsorption processes, and so on, are taken into account; the time-constant dispersion, which can be caused by inhomogeneities in the corroded surface, results in a depression of the semicircle [75, 76]. Adsorption, on the other hand, can reveal a second semicircle at low frequencies [77]. All these effects can occur simultaneously [27], making the interpretation of impedance data rather more difficult [78, 84] (Figure 11).

There is a need for an appropriate model equivalent circuit beyond the existing model standards to remedy that shortcoming. An “appropriate” model is understood not only as a good fit of the impedance data but also as a rational explanation of the underlying corrosion mechanism. Moreover, the requirement of sophisticated AC frequency generator and analyzer and the time needed to acquire the complete impedance diagram (particularly in the range of low frequency) impose a serious limitation in real-time corrosion monitoring applications. Other disadvantages include a priori knowledge of the Tafel parameters in order to convert the polarization resistance into a corrosion rate and the fact that it is too difficult to detect and monitor localized corrosion, even if such applications have been explored [85, 86].
1.4.3. Illustrative examples of the application of EIS in corrosion and tribocorrosion systems
Attempts were made to use the EIS technique in corrosion and corrosion-wear monitoring of Fe-31% Ni electrode immersed in 0.5 M H2SO4 [87]. The corresponding Nyquist impedance diagrams were recorded at an anodic potential of −675 mV/SSE (+100 mV/open-circuit potential) before and during sliding-corrosion as shown in Figure 12. At this potential, the prevailing reaction is dissolution. At high frequency, under free corrosion and unloaded conditions, the capacitive arc reveals the influence of the dielectric properties of the electrochemical double layer and the charge transfer due to electrochemical reactions. Under sliding conditions, the size of the capacitive arc increases, suggesting an increase in the transfer resistance and a decrease in the reactivity of the surface, consistent with the effect of mechanical straining of the worn surface. At low frequency, however, the inductive arc indicates the relaxation of the surface concentration of adsorbed intermediate species involved in the dissolution mechanism. Under corrosion-wear conditions, the kinetics of the dissolution process is apparently modified, as revealed by the second inductive loop in the diagram. Given that not all of these investigations have been concluded, a detailed explanation is not straightforward, and further research is recommended. Although these impedance measurements provide a convenient way to study the mechanism of electrochemical reactions involved in tribocorrosion processes, still the interpretation of impedance records during sliding-corrosion experiments is rather difficult because of the heterogeneous surface-state condition. Actually, a nonuniform distribution of the electrochemical impedance on the steel surface must be taken into account. The action of friction can be analyzed thoroughly if this distribution is known. Equivalent electrical circuit models or finite element models could be used to obtain impedance distributions and to calculate the overall impedance.

2. Comparison of the techniques for the assessment of corrosion rate
The transposition of the foregoing electrochemical techniques to corrosion situations is illustrated in [63] for the assessment of corrosion rate. The results presented in Table 1 summarize the data generated by the different techniques for Fe electrodes in 0.5 M H2SO4 under well-controlled conditions and their corresponding corrosion current densities, resistances, and required parameters which determined those data.
| Techniques | Parameters | ||||||
|---|---|---|---|---|---|---|---|
| ba [mV decade−1] | bc [mV decade−1] | icorr [A cm−2] | CDL[μF cm−2] | RP [Ωcm2] | RΩ [Ωcm2] | Rn [Ωcm2] | |
| Linear polarization (LPR) | — | — | 1.4 × 10−4 | — | 80 | — | — |
| Tafel extrapolation | 34 | 114 | 1.8 × 10−4 Cathodic extrap. | — | — | — | — |
| 7 × 10−5 Anodic extrap. | |||||||
| Electrochemical impedance (EIS) | — | — | 1.1 × 10−4 | 333 (Bode) 116 (Nyquist) 84 (fit) | 98 | 0.3 | — |
| Electrochemical noise (ENA) | — | — | — | — | — | — | 20 –40 |
Table 1.
Data outcomes determined by different electrochemical techniques on Fe in 0.5 M H2SO4.
Reproduced from [63] with permission from Wiley Online Library.
All these techniques monitor the electrode response following the stimulation by a potential variation in time or frequency domain with the exception of the electrochemical noise analysis technique. The extent of the potential stimulation and the current response decreases in the order from Tafel extrapolation method, linear polarization, EIS, to electrochemical noise. Each of these techniques provides the necessary information for a given corroding system, and there are trade-offs involved in the comparative decision of which is the best to use.
Author: Abdenacer Berradja