Click here to see more posts about NMR
Only 15$ for interpretation of your NMR spectrum Payment Upon Completion Send your results...
Nuclear Magnetic Resonance (NMR) interpretation plays a pivotal role in molecular identifications. As interpreting NMR spectra, the structure of an unknown compound, as well as known structures, can be assigned by several factors such as chemical shift, spin multiplicity, coupling constants, and integration. This Module focuses on the most important 1H and 13C NMR spectra to find out structure even though there are various kinds of NMR spectra such as 14N, 19F, and 31P. NMR spectrum shows that x- axis is chemical shift in ppm. It also contains integral areas, splitting pattern, and coupling constant.

Strategy for Solving Structure
Here is the general strategy for solving structure with NMR:
- Molecular formula is determined by chemical analysis such as elementary analysis
- Double-bond equivalent (also known as Degree of Unsaturation) is calculated by a simple equation to estimate the number of the multiple bonds and rings. It assumes that oxygen (O) and sulfur (S) are ignored and halogen (Cl, Br) and nitrogen is replaced by CH. The resulting empirical formula is CaHb
.jpg?revision=1)
- Structure fragmentation is determined by chemical shift, spin multiplicity, integral (peak area), and coupling constants (1J1J, 2J2J)
- Molecular skeleton is built up using 2-dimensional NMR spectroscopy.
- Relative configuration is predicted by coupling constant (3J).
1H NMR
Chemical Shift
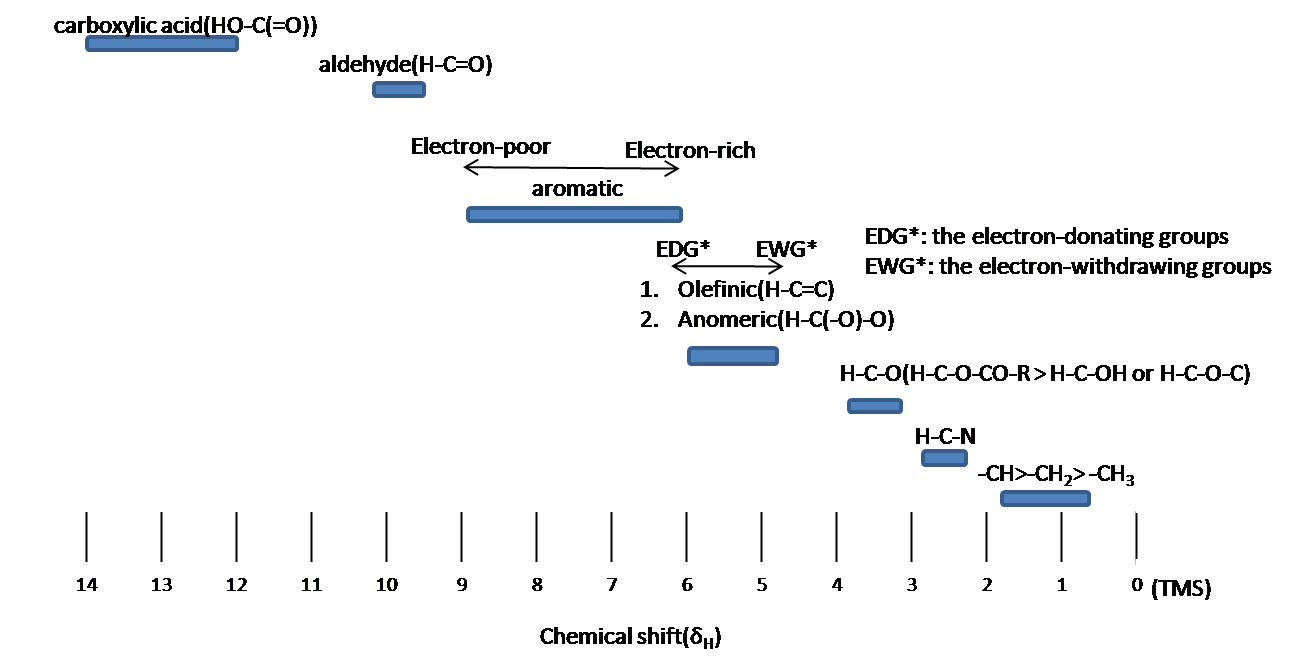
Chemical shift is associated with the Larmor frequency of a nuclear spin to its chemical environment. Tetramethylsilane (TMS, (CH3)4Si(CH3)4Si) is generally used as an internal standard to determine chemical shift of compounds: δTMS=0 ppm. In other words, frequencies for chemicals are measured for a 1H or 13C nucleus of a sample from the 1H or 13C resonance of TMS. It is important to understand trend of chemical shift in terms of NMR interpretation. The proton NMR chemical shift is affect by nearness to electronegative atoms (O, N, halogen.) and unsaturated groups (C=C,C=O, aromatic). Electronegative groups move to the down field (left; increase in ppm). Unsaturated groups shift to downfield (left) when affecting nucleus is in the plane of the unsaturation, but reverse shift takes place in the regions above and below this plane. 1H chemical shift play a role in identifying many functional groups. Figure 11. indicates important example to figure out the functional groups.
Chemical equivalence
Protons with Chemical equivalence has the same chemical shift due to symmetry within molecule (CH3COCH3CH3COCH3) or fast rotation around single bond (-CH3; methyl groups).
Spin-Spin Splitting
Spin-Spin splitting means that an absorbing peak is split by more than one “neighbor” proton. Splitting signals are separated to J Hz, where is called the coupling constant. The spitting is a very essential part to obtain exact information about the number of the neighboring protons. The maximum of distance for splitting is three bonds. Chemical equivalent protons do not result in spin-spin splitting. When a proton splits, the proton’s chemical shift is determined in the center of the splitting lines.
Spin Multiplicity (Splitting pattern)
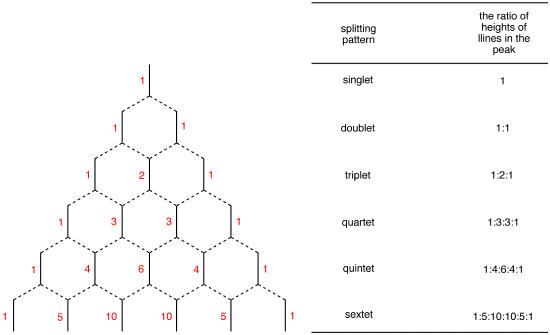
Spin Multiplicity plays a role in determining the number of neighboring protons. Here is a multiplicity rules: In case of AmBnAmBn system, the multiplicity rule is that Nuclei of BB element produce a splitting the AA signal into nB+1nB+1 lines. The general formula which applies to all nuclei is 2nI+12nI+1, where II is the spin quantum number of the coupled element. The relative intensities of the each lines are given by the coefficients of the Pascal’s triangle (Figure 22).
First-order splitting pattern
The chemical shift difference in Hertz between coupled protons in Hertz is much larger than the JJ coupling constant:ΔνJ≥8(1)(1)ΔνJ≥8
Where ΔνΔν is the difference of chemical shift. In other word, the proton is only coupled to other protons that are far away in chemical shift. The spectrum is called first-order spectrum. The splitting pattern depends on the magnetic field. The second-order splitting at the lower field can be resolved into first-order splitting pattern at the high field. The first-order splitting pattern is allowed to multiplicity rule (N+1) and Pascal’s triangle to determine splitting pattern and intensity distribution.
Example 11
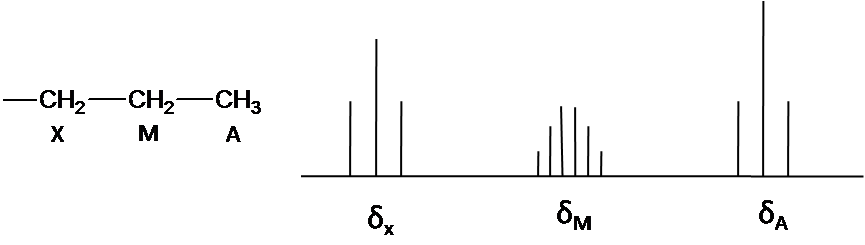
The note is that structure system is A3M2X2. Ha and Hx has the triplet pattern by Hm because of N+1 rule. The signal of Hm is split into six peaks by Hx and Ha(Figure3) The First order pattern easily is predicted due to separation with equal splitting pattern.
High-order splitting pattern
High-order splitting pattern takes place when chemical shift difference in Hertz is much less or the same that order of magnitude as the j coupling.ΔvJ≤10(2)(2)ΔvJ≤10
The second order pattern is observed as leaning of a classical pattern: the inner peaks are taller and the outer peaks are shorter in case of AB system (Figure 44). This is called the roof effect.
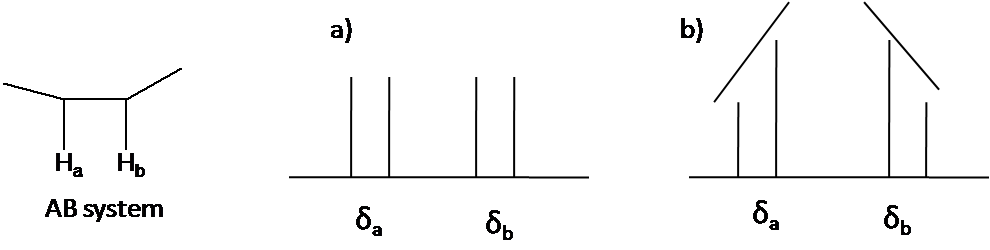
Here is other system as an example: A2B2 (Figure 55). The two triplet incline toward each other. Outer lines of the triplet are less than 1 in relative area and the inner lines are more than 1. The center lines have relative area 2.
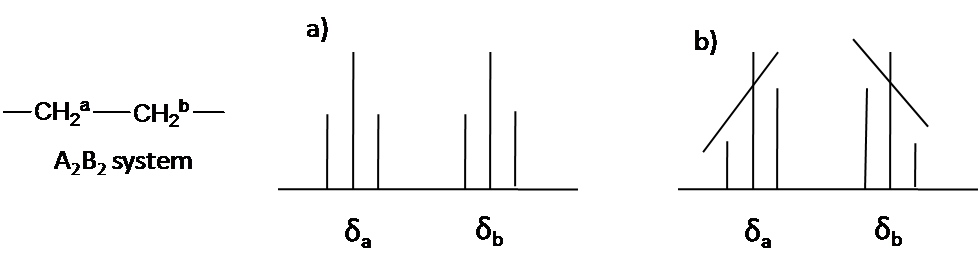
Coupling constant (J Value)
Coupling constant is the strength of the spin-spin splitting interaction and the distance between the split lines. The value of distance is equal or different depending on the coupled nuclei. The coupling constants reflect the bonding environments of the coupled nuclei. Coupling constant is classified by the number of bonds:
Geminal proton-proton coupling (2JHH)
Germinal coupling generates through two bonds (Figure 66). Two proton having geminal coupling are not chemically equivalent. This coupling ranges from -20 to 40 Hz. 2JHHdepends on hybridization of carbon atom and the bond angle and the substituent such as electronegative atoms. When S-character is increased, Geminal coupling constant is increased: 2Jsp1>2Jsp2>2Jsp3 The bond angle(HCH) gives rise to change 2JHH value and depend on the strain of the ring in the cyclic systems. Geminal coupling constant determines ring size. When bond angle is decreased, ring size is decreased so that geminal coupling constant is more positive. If a atom is replace to an electronegative atom, Geminal coupling constant move to positive value.

Vicinal proton-proton coupling (3JHH)
Vicinal coupling occurs though three bonds (Figure 77.). The Vicinal coupling is the most useful information of dihedral angle, leading to stereochemistry and conformation of molecules. Vicinal coupling constant always has the positive value and is affected by the dihedral angle (?;HCCH), the valence angle (?; HCC), the bond length of carbon-carbon, and the effects of electronegative atoms. Vicinal coupling constant depending on the dihedral angle (Figure 88) is given by the Karplus equation.3J=7.0−0.5cosϕ+4.5cos2ϕ(3)(3)3J=7.0−0.5cosϕ+4.5cos2ϕ
When ? is the 90o, vicinal coupling constant is zero. In addition, vicinal coupling constant ranges from 8 to 10 Hz at the and ?=180o, where ?=0o and ?=180o means that the coupled protons have cis and trans configuration, respectively.

The valence angle(?;Figure 88) also causes change of 3JHH value. Valence angle is related with ring size. Typically, when the valence angle decreases, the coupling constant reduces. The distance between the carbons atoms gives influences to vicinal coupling constant

The coupling constant increases with the decrease of bond length. Electronegative atoms affect vicinal coupling constants so that electronegative atoms decrease the vicinal coupling constants.
Integral
Integral is referred to integrated peak area of 1H signals. The intensity is directly proportionally to the number of hydrogen.
13C NMR
Chemical Shift
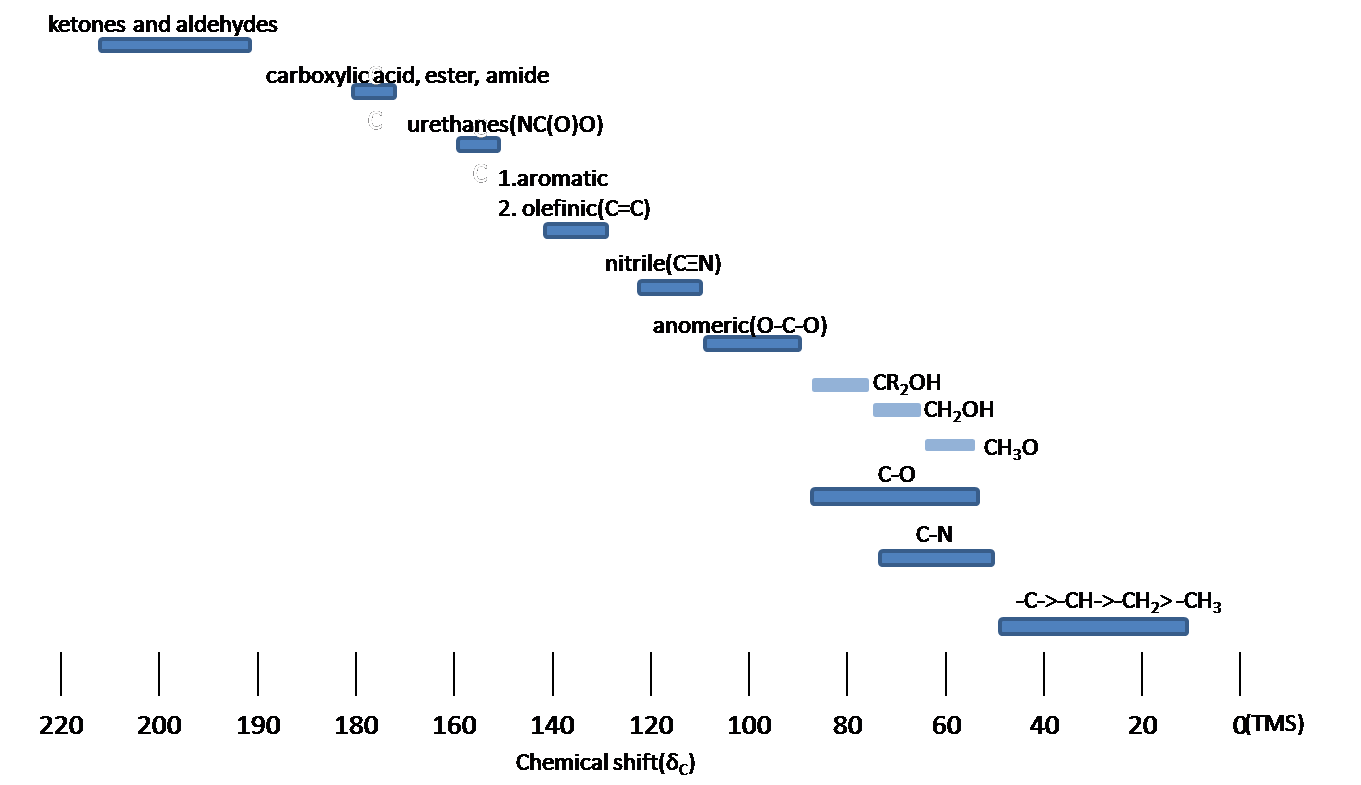
Spin-Spin splitting
Comparing the 1H NMR, there is a big difference thing in the 13C NMR. The 13C- 13 C spin-spin splitting rarely exit between adjacent carbons because 13C is naturally lower abundant (1.1%)
- 13C-1H Spin coupling: 13C-1H Spin coupling provides useful information about the number of protons attached a carbon atom. In case of one bond coupling (1JCH), -CH, -CH2, and CH3 have respectively doublet, triplet, quartets for the 13C resonances in the spectrum. However, 13C-1H Spin coupling has an disadvantage for 13C spectrum interpretation. 13C-1H Spin coupling is hard to analyze and reveal structure due to a forest of overlapping peaks that result from 100% abundance of 1H.
- Decoupling: Decoupling is the process of removing 13C-1H coupling interaction to simplify a spectrum and identify which pair of nuclei is involved in the J coupling. The decoupling 13C spectra shows only one peak(singlet) for each unique carbon in the molecule(Figure 1010.). Decoupling is performed by irradiating at the frequency of one proton with continuous low-power RF.

- Distortionless enhancement by polarization transfer (DEPT): DEPT is used for distinguishing between a CH3 group, a CH2 group, and a CH group. The proton pulse is set at 45o, 90o, or 135o in the three separate experiments. The different pulses depend on the number of protons attached to a carbon atom. Figure 1111. is an example about DEPT spectrum.

2-dimensional NMR spectroscopy (COSY)
COSY stands for COrrelation SpectroscopY. COSY spectrum is more useful information about what is being correlated.
1H-1H COSY (COrrelation SpectroscopY)
1H-1H COSY is used for clearly indicate correlation with coupled protons. A point of entry into a COSY spectrum is one of the keys to predict information from it successfully. Relation of Coupling protons is determined by cross peaks(correlation peaks) and in the COSY spectrum. In other words, Diagonal peaks by lines ar e coupled to each other. Figure 1212 indicates that there are correlation peaks between proton H1 and H2 as well as between H2 and H4. This means the H2 coupled to H1 and H4.

1H-13C COSY (HETCOR)
1H-13C COSY is the heteronuclear correlation spectroscopy. The HETCOR spectrum is correlated 13C nuclei with directly attached protons. 1H-13C coupling is one bond. The cross peaks mean correlation between a proton and a carbon (Figure 1313). If a line does not have cross peak, this means that this carbon atoms has no attached proton (e.g. a quaternary carbon atom)

References
- Balc*, M., Basic p1 sH- and p13 sC-NMR spectroscopy. 1st ed.; Elsevier: Amsterdam ; Boston, 2005; p xii, 427.
- Breitmaier, E., Structure elucidation by NMR in organic chemistry : a practical guide. 3rd rev. ed.; Wiley: Chichester, West Sussex, England, 2002; p xii, 258.
- Jacobsen, N. E., NMR spectroscopy explained : simplified theory, applications and examples for organic chemistry and structural biology. Wiley-Interscience: Hoboken, N.J., 2007; p xv, 668.
- Silverstein, R. M.; Webster, F. X., Spectrometric identification of organic compounds. 6th ed.; Wiley: New York, 1998; p xiv, 482.
Outside Links
- NMRShiftDB: a Free web database for NMR data : nmrshiftdb.chemie.uni-mainz.de/nmrshiftdb
- NMR database from ACD/LAbs : www.acdlabs.com/products/spec_lab/exp_spectra/spec_libraries/aldrich.html
- NMR database from John Crerar Library : http://crerar.typepad.com/crerar_lib…h_ir_nmr_.html
Problems
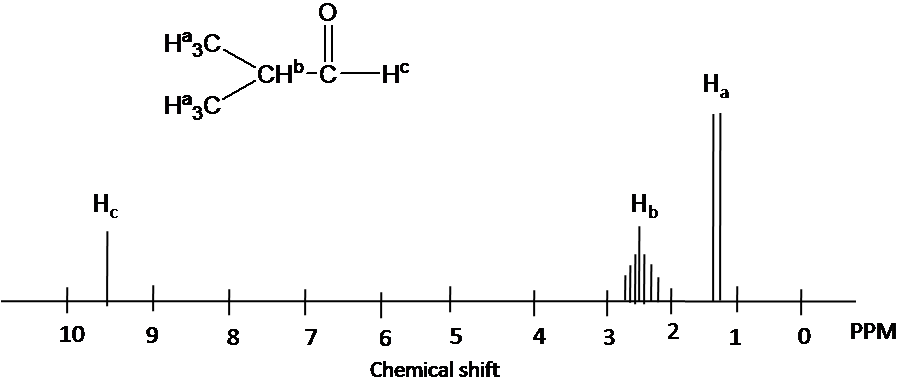
Draw the 1H NMR spectrum for 2-Hydroxypropane in CDCl3. Assume sufficient resolution to provide a first-order spectrum and ignore vicinal proton-proton coupling(3JHH)
Solution
1) the structure of 2-hydoroxyporpane is drawn

Figure out which protons are chemically equivalent, i.e., two methyl (-CH3) groups are chemical equivalent.
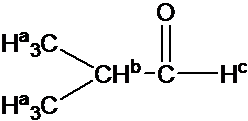
4) Splitting pattern is determined by (N+1) rule: Ha is split into two peaks by Hb(#of proton=1). Hb has the septet pattern by Ha (#of proton=6). Hc has one peak.(Note that Hc has doublet pattern by Hb due to vicinal proton-proton coupling.)